[title]ALD简述[/title]
疾病档案
ICD-10: 8A44.1
OMIM: 300100
ORPHA: 43
致病基因: ABCD1(位于xq28)
肾上腺脑白质营养不良,简称ALD,是一种罕见的遗传代谢病,是由位于X染色体上的ABCD1基因突变引起的,该突变导致极长链脂肪酸(VLCFA)无法正常代谢,从而造成神经系统中白质的逐渐丧失和肾上腺皮质的退化。脑型(cALD)的患者大部分在较短时间内完全瘫痪,甚至死亡。
[title]患病率[/title]
95%的患者为男性,而女性多为杂合子,属于疾病基因突变的携带者。男性ALD的发病率为1/21000~1/15500,而男性ALD 和女性杂合子携带者的共同发病率为1/17000。
[title]遗传机制[/title]
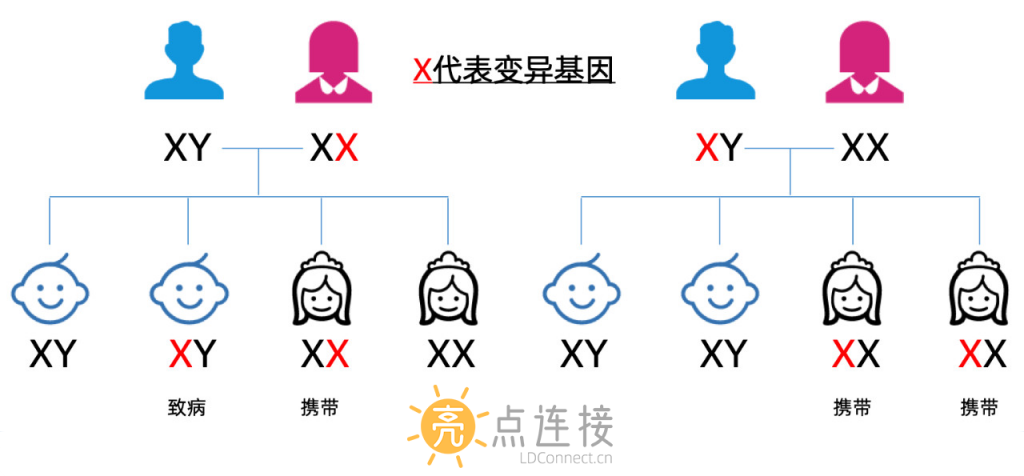
X连锁隐性遗传,男性只有一个X染色体,所以只要获得包含突变基因的X染色体,几乎100%发病。
而女性因为有两个X染色体,一个X染色体包含变异基因,一般不会发病,而成为突变ABCD1基因携带者,在生育后代时,有50%的几率将突变的ABCD1基因遗传给孩子。
[title]发病分型和症状[/title]
儿童脑型
是ALD的常见类型,占所有ALD患者35%。又称CCALD。多为4-8岁男孩发病,大脑中枢神经脱髓鞘病变。
初期表现为注意力不集中、记忆力减退、学习困难、步态不稳、行为异常,逐步出现认知、行为、言语、视力、听力和运动功能的进行性损害,并通常在六个月至两年内导致完全残疾。在神经系统症状出现时,大多数人的肾上腺皮质功能已经受损。预后很差。从发病开始到死亡,大多不超过36个月。死因一般是中枢性呼吸衰竭、脑疝、感染等。
青少年脑型
占比5%左右的类型,多为10-20岁男孩发病,症状类似CCALD,但进展稍慢。
成人脊髓型
是ALD的常见类型,约占27%,又称AMN(肾上腺脊髓神经病),大部分成年男性起病都是这种类型。20-50岁发病,主要累及脊髓白质,周围神经病变较轻,无炎症性损伤。表现为下肢进行性痉挛性瘫痪、括约肌功能紊乱、性功能障碍以及肾上腺皮质功能受损。AMN 进展较慢,初期很容易被误诊。本站长就是这种类型。
成人脑型
少部分成年男性患者(3%左右),起病就影响脑部,同时有35%左右的AMN患者随着疾病进展会出现大脑脱髓鞘的情况,影响大脑以后,疾病进展也较快。
艾迪生型
占比10~14%的类型,在2岁以后出现原发性肾上腺皮质功能不全症状,包括皮肤色素沉着、虚弱无力、多汗、嗜盐,伴有呕吐、腹泻甚至低血压晕厥等,无神经受累症状。很容易误诊为先天性肾上腺皮质功能减退症。该类型后期仍有可能发展为CCALD或者AMN。
女性杂合子型
超过20%的女性携带者在中年或以后发展为轻度至中度痉挛性轻瘫,在60岁后,65%的杂合子患者会出现AMN临床表现,但症状轻微。很少出现脑部症状、周围神经病及肾上腺皮质功能减退。
无症状型
通过检查发现血极长链脂肪酸(VLCFA)升高或ABCD1基因存在突变,但患者无相应的临床症状。
[title]诊断[/title]
- 通过基因检测确定ABCD1基因的突变情况是最重要的确诊手段
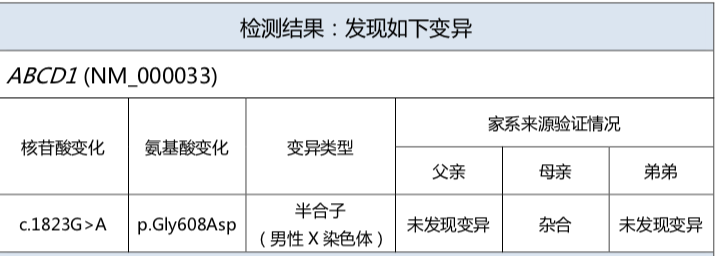
- 极长链脂肪酸水平测定、肾上腺皮质醇激素测定是主要生化检测手段。VLCFA增高是诊断疾病的主要生化指标,见于几乎所有男性患者及85%的女性携带者。VLCFA 升高水平与病情的严重程度无关。检测羊膜细胞和绒毛膜细胞中VLCFA,可用于产前诊断;需要注意,10%~15%的女性杂合子携带者的VLCFA 水平并不高。
- 头颅MRI也能帮助医生确定病情发展
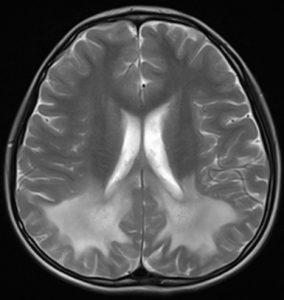 其特征性的影像学改变是双侧顶枕区白质内对称分布的蝴蝶状长T1、T2 信号,增强检查可见病灶周围呈镶边样强化;如果病变进一步进展,病灶的累及范围可以由脑组织后部向前部扩展。
其特征性的影像学改变是双侧顶枕区白质内对称分布的蝴蝶状长T1、T2 信号,增强检查可见病灶周围呈镶边样强化;如果病变进一步进展,病灶的累及范围可以由脑组织后部向前部扩展。
[title]治疗[/title]
ALD不能被治愈,但可以治疗,也可以通过产前干预(三代试管婴儿或者孕期胎儿基因检测)来预防。
[title]视频[/title]
如小程序内看不到视频,请点左下角“阅读原文”。或使用浏览器访问网页:https://ldconnect.cn/ald/ald-intro.html
[title]参考资料[/title]
[1] Genereview: https://www.ncbi.nlm.nih.gov/books/NBK1315/
微信扫描下方的二维码阅读本文


