我是轩飞,今年43岁,一名“资深”互联网从业者,发量可鉴。
关注罕见病领域,并非偶然,始于2019年7月。
2019年7月初,当我拿到基因检测报告,听医生描述我的问题的时候,当时的感受是很不真实的。花了十三年,我终于找到了影响自身健康的真正原因——一种遗传代谢病——肾上腺脑白质营养不良,简称ALD,然而这种疾病并没有有效的治疗方案,医生说这些术语的时候,我大脑一片空白,这种恍惚持续了好几天。
回过神来,我加了病友群,花了十来天上网找了关于ALD的近期国内外医学论文,看了一部豆瓣评分8.5的影片《洛伦佐的油》。
这是一种可怕的罕见病,十万分之一的发病率,95%的患者是男性。我身体里两万多个基因中,有一个叫做ABCD1的基因发生了变异,无法产生一种叫做ALDP的蛋白,这种蛋白能帮助「极长链脂肪酸」的氧化代谢,就像没有胰岛素,血糖无法代谢一样,没有ALD蛋白,极长链脂肪酸就无法正常代谢。于是血液里的极长链脂肪酸浓度就越来越高,随着血液循环沉积在器官上,损害肾上腺、神经的髓鞘、大脑等等。我是ALD成人发病,渐进性的脊髓受损造成了我的主要问题。随着时间推移,问题会越来越严重,这些年我身体的各种变化,都变得有据可循。
过往数据表明,45%的ALD成年脊髓型患者最终大脑也会受损,也许有一天我会无法行走,甚至无法再用键盘敲出这样的文字...
你可能觉得我很不幸,其实恰恰相反,我是ALD患者中,非常幸运的那一个!
-
如果我在童年发病(CCALD),我可能活不过36个月;
-
如果我在青少年时期发病,我可能无法完成学业,结婚生子,成家立业;
-
-
如果我生了女儿,她将来的婚恋和生育,将面临艰难的选择...
时间回到我27岁那年的夏天,我打篮球崴了脚,这不是第一次崴脚,不同寻常的是,消肿后,恢复锻炼时,我发现跑步时双腿像灌了铅一样,跑50米就无法再抬起脚来,也许这次只是需要更多时间恢复?一年后我有了一对双胞胎儿子,又过三年,孩子们已经跑得飞快,而我却已经完全不能跑步了。心里有一个声音说:「一定要好起来,等孩子大些,就可以一起跑步了!」。
我开始四处求医,辗转不同的医院和科室,几乎每年都要做核磁共振,吃药、针灸也成了家常便饭,14年和16年分别做了一次骨科手术,然而奇迹并没有发生。孩子越来越大,我却越来越像一个蹒跚学步的孩子。神经内科医生认为我有某种渐行型的运动神经问题,原因未明!
已经认命的我,在2019年5月底,按神经内科医生建议,做了一次基因检测,7月初报告出来,如前文所述,求医十三年,我终于确诊了。
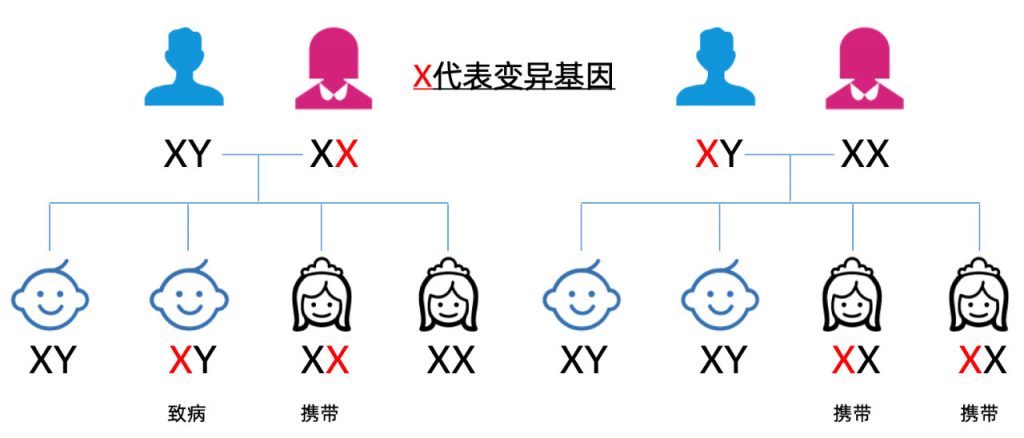
ALD是一种X性染色体隐性遗传病,我的变异基因遗传自我的母亲,姐姐也有50%的几率获得这个基因。8月初,我做足了功课,我才和姐姐说起了这件事。很快,姐姐做了基因检测,报告显示,她也是ABCD1致病基因的携带者,这意味着她的一儿一女都有50%的机率遗传致病基因,如果我的小外甥有这个基因的话,99%会发病!!!
两个孩子的血样送到基因检测公司后,可以想象,十几天的等待,对于姐姐和姐夫来说,是怎样的煎熬...
报告出来了!谢天谢地,幸运女神又一次和我们家站在了一起,外甥的ABCD1基因正常,而外甥女遗传了致病基因,也是一名携带者,未来的生育需要选择。虽不完美,但也足够幸运!
ALD的变异基因携带率为1/16800,而发病率为1/100000,基本上携带变异基因的女性在35岁以前不会有任何症状,如果一个家族中没有出现我这样的“首证者”,没有人会在孕前做相关的检查。不知情的女性携带者生育下一代,变异基因有50%的几率将被遗传。如果生育男孩,当儿子发病,病魔一点一点的夺走孩子生命时,母亲的心痛,难以言表...
类似ALD这样的脑白质营养不良类罕见遗传病还有很多种,都面临和ALD一样无药可医的情况。
建立这个网站,希望能连接更多病友,推动相关医药研究,也让病友少走弯路,避免“病急乱投医”。
作为一个罕见病人,上天给到我的使命,我将尽力履行。
希望可以连接所有的脑白质营养不良病友,毕竟唯有病友才能真正关爱彼此。
微信扫描下方的二维码阅读本文