[title]MLD简述[/title]
异染性脑白质营养不良,简称MLD,是一种罕见的常染色体隐性遗传代谢病,是由ARSA基因(22q13.33)突变引起芳基硫酸酯酶A 缺乏从而无法正常代谢硫苷脂,过量硫苷脂会破坏生成髓磷脂的细胞,引发脱髓鞘,进而造成神经系统中白质的逐渐丧失。
芳基硫酸酯酶A位于溶酶体中,因此MLD也被归为“溶酶体储积病”。
另有少量病例是PSAP基因(10q22.1)突变引起的saposin B蛋白质缺乏,这种蛋白质可以激活芳基硫酸酯酶A 共同分解硫苷脂,如果缺乏也会带来ARSA酶缺乏一样的问题。
[title]患病率[/title]
MLD的具体患病率未知,但新生儿患病率估计在1/160000~1/40000之间。据认为MLD基因突变的携带率约为1/100。
[title]遗传机制[/title]
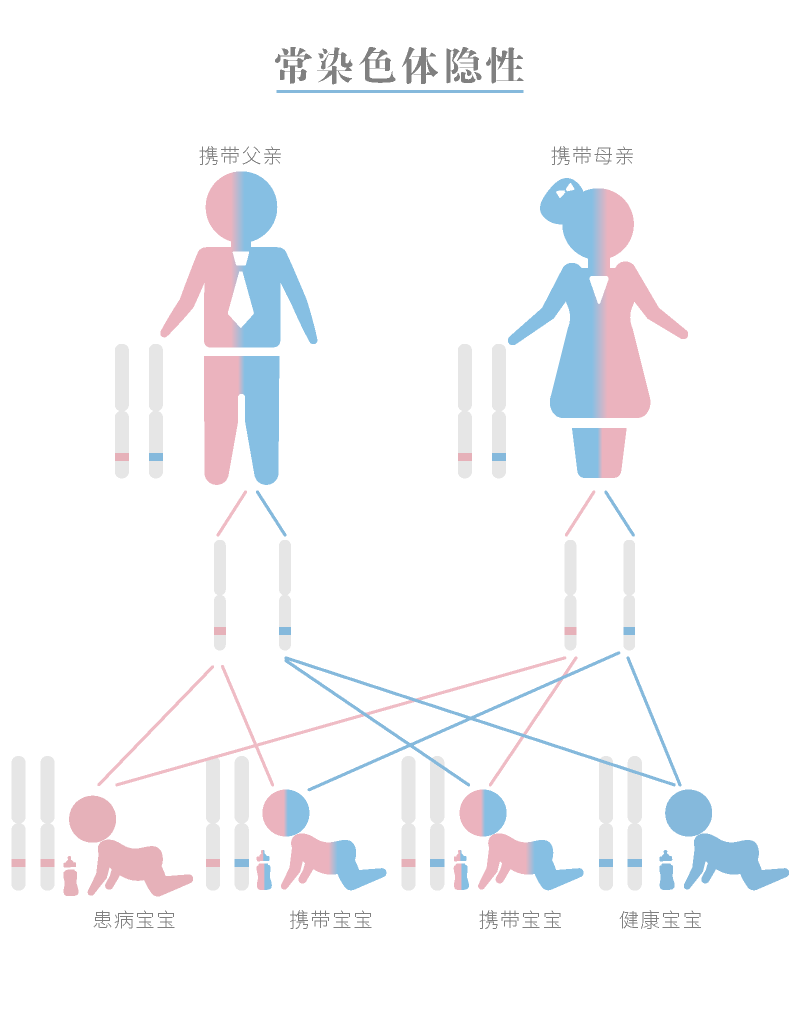
常染色隐性遗传,携带一个变异基因并不会发病,如果父母双方都携带变异基因,则子女有25%的几率患病。
[title]发病分型和症状[/title]
晚期婴儿型
最常见形式(50%-60%),常在出生后两年内发病,患病的孩子发育迟缓,失去语言能力和行走障碍。随着疾病的恶化,肌张力通常先下降,然后增加到僵硬的程度,患者通常不能存活到童年以后。
少年型
20-30%的患者,发病发生在4岁至青春期之间。最初征兆可能是行为问题以及学习障碍,进展比晚期婴儿型要慢,患者一般可以存活20年左右。
成人型
约15%至20%的患者,症状出现在青少年时期或以后,通常会出现诸如酗酒,药物滥用、学习或工作障碍等行为问题。并可能会出现精神病症状,例如妄想或幻觉,患者一般可以存活20至30年,期间,病程发展可能相对稳定,有时也快速发展。
[title]诊断[/title]
- 针对RASA和PASA基因突变情况的基因检测可以作为确诊依据
- 头颅MRI可以帮助医生判断是否为MLD,但不足以作为确诊依据
- 血液ARSA酶水平检查和血尿硫苷脂检查是MLD最合适的生化检查

[title]治疗[/title]
- 对于未发病或者症状轻微的患者可以考虑干细胞移植
- 对于症状明显的患者主要是缓解神经功能症状
- 有多个进行中的基因治疗临床实验:主要是慢病毒载体或自体干细胞基因修饰回输。
[title]视频[/title]
[title]参考资料[/title]
[1] Genetics Home Reference: https://ghr.nlm.nih.gov/condition/metachromatic-leukodystrophy
[2] Wikipedia: https://en.wikipedia.org/wiki/Metachromatic_leukodystrophy
[4] Clinicaltrials: https://www.clinicaltrials.gov/ct2/results?cond=MLD&map_cntry=CN&draw=2&rank=1#rowId0
微信扫描下方的二维码阅读本文


