[title]亚历山大病简述[/title]
ICD-10: E75.2
OMIM: 203450
ORPHA: 58
致病基因: GFAP
亚历山大病(Alexander disease),简称ALX或AxD,是一种非常罕见的遗传代谢病,约95%是由17号染色体上的GFAP基因(17q21.31)突变造成神经胶质原纤维酸性蛋白(GFAP)异常,影响髓磷脂生成,引起脱髓鞘,破坏脑白质,并在大脑和脊髓神经中形成一种纤维状的嗜酸性粒细胞沉积,称为罗森塔尔纤维(Rosenthal fiber)。
疾病命名来自于1949年发现这种疾病的澳大利亚病理学家W. Stewart Alexander博士。
[title]患病率[/title]
亚历山大病非常罕见,全球已报道过的仅500多例。
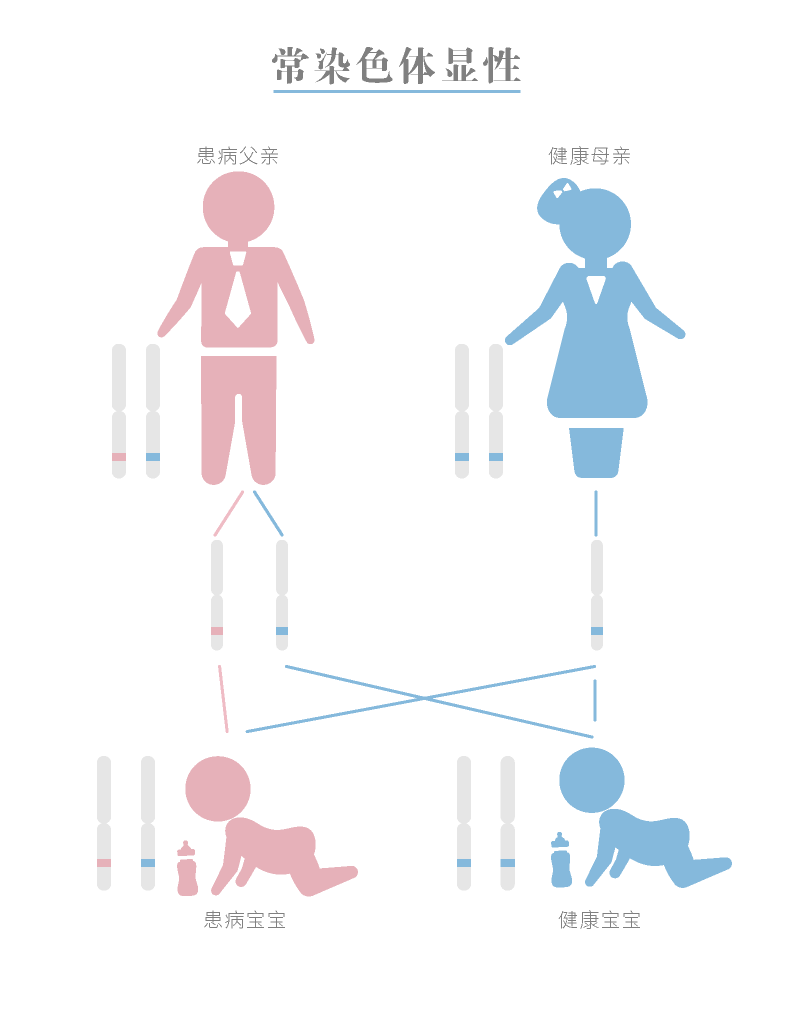
[title]遗传机制[/title]
常染色显性遗传,父母双方的任何一方患病,子女都有50%的几率患病。但大部分亚历山大病是新发的突变,而非遗传自父母。
[title]发病分型和症状[/title]
新生儿型
极少数患者在出生后一个月内发病,症状包括癫痫发作、脑积水、严重的运动和智力残疾以及脑脊液蛋白浓度升高。MRI显示包括基底神经节和小脑的严重白质异常。通常在两年内导致严重的残疾甚至死亡。
婴儿型
约42%的患者,在2岁内发病,通常表现为进行性精神运动发育迟缓、大头畸形、额突和癫痫发作。另外还可能发生反射亢进、锥体束征、共济失调和脑积水。患病儿童可以存活数周至数年。
幼年型
约22%的患者,在4至10岁之间发病,偶尔出现在15岁左右,症状包括延髓/假性延髓体征、共济失调、智力逐渐丧失、癫痫发作、大头畸形以及呼吸问题。存活时间从十几岁到二、三十岁不等。
成人型
约33%的患者在成人后发病,可能出现延髓/假性延髓体征、锥体束体征、小脑体征、自主神经失调、睡眠障碍、步态障碍、偏瘫或四肢瘫痪、癫痫和复视。发病后生存期从几年到几十年不等。
[title]诊断[/title]
- 针对GFAP基因突变情况的基因检测可以作为确诊依据,但是阴性结果也不能排除患病可能
- 头颅MRI可以帮助医生判断,但需要排除其它相关LD疾病
- 脑活检确定罗森塔尔纤维的存在也是重要检查方式,但不可以作为确诊依据
[title]参考资料[/title]
微信扫描下方的二维码阅读本文


